V
主页
Ni表面吸附H模型构建 | 【朱老师讲DFT计算】
发布人
Ni表面吸附H模型,吸附位点,层间距 【计算入门】VASP计算九大专题培训:晶体、二维材料、催化、电池、钙钛矿、单原子、吸附、半导体、缺陷计算等!链接:https://mp.weixin.qq.com/s/SzDIroX_T7E5ZSSwO4zqfw DFT计算培训、代算请咨询:小硕131-2872-3011 点击链接咨询:https://dwz.cn/oGtmDZFr
打开封面
下载高清视频
观看高清视频
视频下载器
Ni表面吸附H结合能计算 | 【朱老师讲DFT计算】
Ni表面吸附H2O结构优化计算 | 【朱老师讲DFT计算】
VASP公开课——一节课入门催化与吸附计算
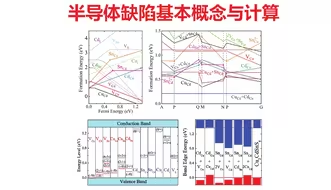
缺陷计算:半导体缺陷基本概念与计算 | 【华算科技朱老师讲DFT-VASP】
MoS2态密度分析 | 【朱老师讲DFT计算】
Ni晶体结构优化计算 | 【朱老师讲DFT计算】
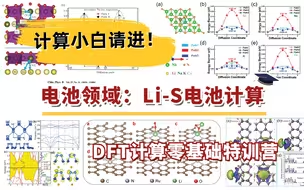
电池领域:Li-S电池计算 | DFT计算特训营【朱老师讲VASP】
顶刊中的电荷图:差分电荷分布计算与分析
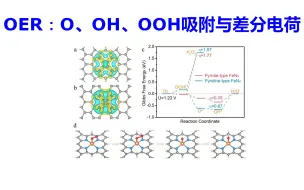
催化OER计算:OER中间体O、OH、OOH吸附与差分电荷 | 【华算科技朱老师讲DFT-VASP】
电催化HER自由能台阶图 |【朱老师讲DFT计算】
吸附结构:吸附位点、吸附构型 | DFT计算特训营【朱老师讲VASP】
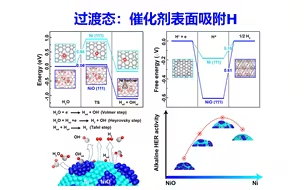
过渡态计算H吸附在催化剂表面、初态、末态 | DFT计算特训营【朱老师讲VASP】
VASP钙钛矿结构分析 | 【朱老师讲DFT计算】
N掺杂C负载Fe单原子吸附H模型构建 | 【朱老师讲DFT计算】
VASP表面吸附与催化计算培训:HER、OER/ORR、NRR、CO2RR、表面性质,吸附能、差分电荷密度、d带中心、自由能、反应过渡态等
Ni表面结构分析 | 【朱老师讲DFT计算】
MoS2负载Fe单原子吸附CO2分子吸附能 | 【朱老师讲DFT计算】
Ni表面吸附H2O结合能 | 【朱老师讲DFT计算】
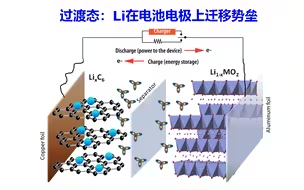
过渡态计算Li在电池电极上迁移路径与势垒 | DFT计算特训营【朱老师讲VASP】
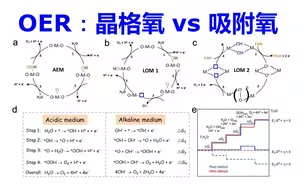
催化OER计算:反应路径晶格氧vs吸附氧机理 | 【华算科技朱老师讲DFT-VASP】
Ni表面吸附H差分电荷密度计算 | 【朱老师讲DFT计算】
Ni表面态密度计算 | 【朱老师讲DFT计算】
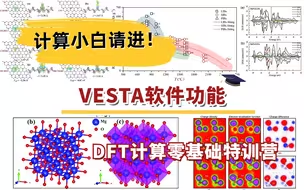
VESTA软件功能:模型构建、理论XRD、电荷密度绘图 | DFT计算特训营【朱老师讲VASP】
QE电荷密度计算 | 【华算科技朱老师讲DFT-QE】
电催化CO2RR自由能台阶图 |【朱老师讲DFT计算】
电荷密度:自洽计算、CHGCAR文件、VESTA作电荷密度图 | DFT计算特训营【朱老师讲VASP】
DFT计算在半导体、催化、电池领域的应用 | 科研话题系列视频第一期 【朱老师讲VASP】
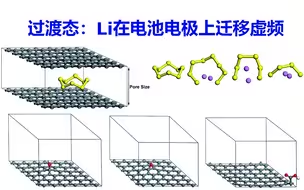
过渡态计算Li在电池电极上迁移、原子振动频率、虚频 | DFT计算特训营【朱老师讲VASP】
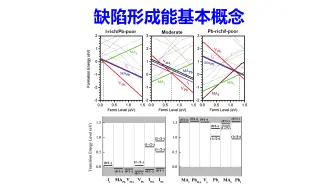
缺陷计算:半导体缺陷形成能基本概念 | 【华算科技朱老师讲DFT-VASP】
N掺杂C负载Fe单原子吸附CO2差分电荷密度 | 【朱老师讲DFT计算】
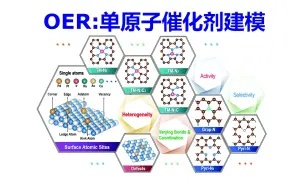
催化OER计算:单原子结构模型构建 | 【华算科技朱老师讲DFT-VASP】
MoS2吸附CO2分子CO2结构优化计算 | 【朱老师讲DFT计算】
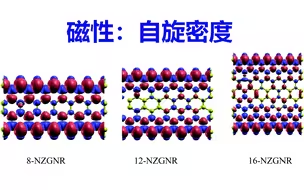
磁性计算:自旋密度计算 | 【华算科技朱老师讲DFT-VASP】
VASP脚本:bader 用于分析原子间的电荷转移 | 【朱老师讲DFT计算】
局域态密度作图与分析 | 态密度计算与应用-DFT计算特训营【朱老师讲VASP】
VASP Li-S电池计算培训:单/双原子、纳米颗粒、晶体表面模型,电子性质,d带中心、多硫化锂吸附于转化、反应过渡态
N掺杂C负载Fe单原子吸附H结构优化 | 【朱老师讲DFT计算】
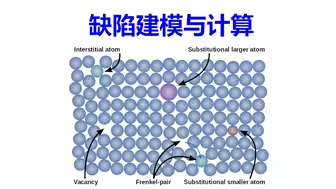
缺陷计算:半导体缺陷建模与计算 | 【华算科技朱老师讲DFT-VASP】
N掺杂C负载Fe单原子吸附NO3模型构建 | 【朱老师讲DFT计算】
电催化NO3RR自由能台阶图 |【朱老师讲DFT计算】