V
主页
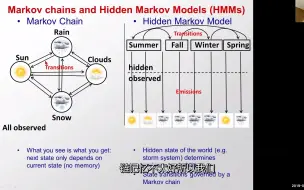
麻省理工学院计算生物学4 Hmms隐马尔可夫模型-2
发布人
本次讲座的大纲: 1.回顾:上次的基础知识和算法 -P(x,π)。Viterbi:π*=argmaxπP(x,π)。正向:P(x) -后验解码:找到最可能的状态πi,所有路径。 2.增加“状态”空间/增加内存 -寻找富含GC的区域与寻找CpG岛 -基因结构GENSCAN,染色质ChromHMM 3.学习(ML训练、Baum-Welch、Viterbi训练) -监督:查找给定标记序列的ei(.)和aij -无监督:仅给定x 注释+参数 4.条件随机场(CRF)和相关性 -不同输入、模型依赖性的HMM限制 -CRF定义。建模依赖于CRF。 -将HMM表示为CRF的特例
打开封面
下载高清视频
观看高清视频
视频下载器
麻省理工学院计算生物学4 Hmms隐马尔可夫模型-1
麻省理工学院计算生物学2序列比对与动态规划
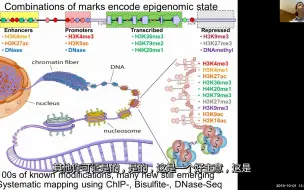
麻省理工计算生物学8:表观基因组学
多组学ln代谢病2023主题演讲
路径富集分析——简单解释
Aucell的单细胞基因集活性
什么是序列比对?
如何解释Gsea结果和绘图
麻省理工学院计算生物学3哈希Blast数据库搜索
麻省理工计算生物学6:基因表达分析之聚类分类
Logistic回归诺模图
使用R包StageWise轻松进行基因组选择
批量bulk测序与单细胞bulk测序的入门指南
一个视频带你看懂RNA的结构、功能和类型
单细胞ATAC-seq分析教程【二】与scRNA-seq的异同
机器学习R语言讲解 -构建和验证统计模型的教程
什么是GRanges以及用法
尼安德特人基因组计划:对人类进化的洞察
R 语言使用clusterProfiler()的途径富集分析教程
未被注意的异质性
基因簇分析
比较基因组学讲座-基因组学导论
单细胞ATAC-seq分析方法【一】测序原理
我们为什么要使用Aws
MIT计算生物学——蛋白质序列的全局比对
如何用Sr图进行基因富集(Go和Kegg途径)分析
单细胞RNA-seq分析方法【十二】反卷积
生物信息历史
TBtool进行GO基因注释,助你了解你的基因功能
人类免疫系统-它是如何工作的!-1
蛋白质结构和功能
使用Singler方法进行单细胞细胞类型自动注释1
生物信息学与计算生物学
生存函数,或风险比的分析
随机森林概述和R演示
什么是基因组测序-2
使用R语言介绍单细胞RNA-Seq数据的差异表达特征及聚类识别
多层数据简介
基因集富集分析(GSEA)
五分钟讲解Statquest Pca主要要点以及概念