V
主页
主流分子可视化软件对原子间成键判断规则
发布人
原子间是否成键判断方式有很多,最为正确的当然是根据本身已知分子的性质,化学环境判定。 在分子模拟领域可视化分子过程中,往往用户并不会直接提供原子正确的连接信息,此时需要可视化软件自行判断。这种情况下不同软件给出可能并非一致。 目前网上已有可参考的较为全面的资料: ①谈谈原子间是否成键的判断问题 (http://sobereva.com/414) ②谈谈VMD可视化程序的连接关系的判断和设置问题 (http://bbs.keinsci.com/thread-16396-1-1.html)
打开封面
下载高清视频
观看高清视频
视频下载器
VMD拓展插件的使用
分子动力学中氢键的计算方法和差异
基于VMD的氧化石墨烯建模工具
使用GROMACS自带工具x2top创建周期性氧化石墨烯拓扑文件
GROMACS杂谈之一: 常用分子力场和适用情况
GROMACS系列教程之一: 小分子过双层磷脂膜PMF的计算流程
gromacs_on_windows
Ovito在GROMACS轨迹分析中的应用
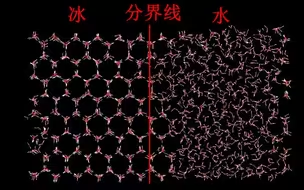
GROMACS模拟水结冰过程
TprParser - 高性能GROMACS输入tpr文件读写插件库
GROMACS中一些特殊的位置限制及用法
使用GROMACS模拟甲烷水合物形成过程
genTop: 快速GROMACS拓扑文件生成工具
Qtgrace/Xmgrace在GROMACS后处理作图中的应用
快速分子系统搭建工具-FastPackMol
pymol_install_on_linux
MolViewer - GROMACS相关的轻量级小程序
分子动力学磷脂膜构建工具--PymixMem
分分钟了解GROMACS 2023新特性
一分半钟看完GROMACS 2024新特性!(重制)
GROMACS模拟的同一体系不同周期性分子解决轨迹处理及可视化
水分子F3/F4序参数计算编程
基于OpenBabel批量产生复杂基团以任意方式接到某个中心基团上
GROMACS 2022 新特性详解
基于TPPMKTOP模拟简单线性聚合物(聚丙烯酰胺)
教大家理解范德华作用2个参数对于计算聚集的影响
官网的Pymol水印移除小工具
How to simulate organic molecules by GROMACS, Ambertools and acpype on Windows
How to extract molecule structure and atomic charges from .tpr file of gromacs
分子动力学AMBER新功能速递(ML/MM)
GROMACS数据作图工具 -- gplt
分子动力学模拟-第12讲-GPUMD介绍(樊哲勇)
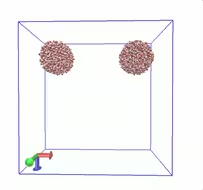
使用GROMACS模拟两水球碰撞过程2
[Materials Studio] Forcite模块 10个热门痛点问题
功能化碳纳米管建模方案
GMXpbsatool_On_Windows
How to use g_mmpbsa on Windows
两种不同力场下较高浓度NaCl溶液模拟:Amber99sb vs KBFF20
GROMACS 2021新特性详解
无限大/小片氧化石墨烯(氧化度20%)未添加限制下的分子动力学模拟形态