V
主页
钙钛矿电子结构:态密度 | DFT计算特训营【朱老师讲VASP】
发布人
培训/代算及公开课课程资料请联系小硕! ✅联系方式:131-2872-3011 钙钛矿材料在太阳能电池、半导体期间、光电器件等方面具有突出的性能,从而受到人们广泛的关注。 模型构建与参数设置是开展DFT计算的准备工作,尤其是有机结构部分,对初学者来说往往具有一定的难度。 本次DFT计算公开课将由华算科技的朱老师给大家介绍钙钛矿材料建模与DFT计算,具体涉及阴离子与阳离子模型、三维钙钛矿模型、二维钙钛矿模型、态密度、HOMO/LUMO、结合能、差分电荷密度等重要性质。 ✅计算模型构建: 1.阴离子与阳离子模型:BF4、BA、MA、FA模型 2.三维模型:含不同阳离子的三维钙钛矿结构 3.二维模型:含不同末端原子的二维钙钛矿模型。 ✅电子性质计算: 1.态密度:包括总态密度、原子轨道分态密度、局域态密度 2.HOMO/LUMO轨道:价带顶和导带底的空间分 ✅吸附性质计算: 1.吸附构型:分子或原子在钙钛矿表面的最稳定吸附构型 2.结合能:描述分子或原子与钙钛矿相互作用强弱 3.差分电荷密度:分子或原子与钙钛矿电子转移情况 朱老师深耕计算领域13年,资深技术手把手带你入门!
打开封面
下载高清视频
观看高清视频
视频下载器
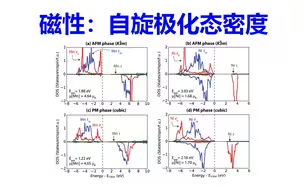
磁性计算:自旋极化态密度计算 | 【华算科技朱老师讲DFT-VASP】
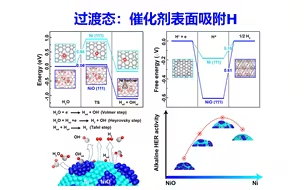
过渡态计算H吸附在催化剂表面、初态、末态 | DFT计算特训营【朱老师讲VASP】
吸附结构:吸附位点、吸附构型 | DFT计算特训营【朱老师讲VASP】
钙钛矿电子结构:HOMO/LUMO | DFT计算特训营【朱老师讲VASP】
VASP钙钛矿结构优化计算 | 【朱老师讲DFT计算】
磁性计算:反铁磁构型能量计算 | 【华算科技朱老师讲DFT-VASP】
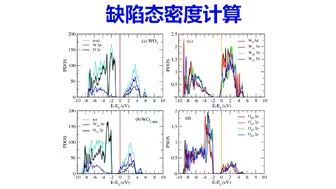
缺陷计算:半导体缺陷态密度计算 | 【华算科技朱老师讲DFT-VASP】
总态密度作图与分析 | 态密度计算与应用-DFT计算特训营【朱老师讲VASP】
能带结构 |【朱老师讲DFT计算】
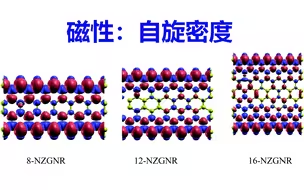
磁性计算:自旋密度计算 | 【华算科技朱老师讲DFT-VASP】
d带中心计算 | 态密度计算与应用-DFT计算特训营【朱老师讲VASP】
电子性质计算:态密度、能带结构、投影电荷密度 | DFT计算在钙钛矿领域的应用
【朱老师讲VASP】纯小白入门DFT计算:Si态密度计算
VASP钙钛矿电荷密度计算 | 【朱老师讲DFT计算】
电子态密度 |【朱老师讲DFT计算】
N掺杂C负载Fe单原子吸附H结合能 | 【朱老师讲DFT计算】
VASP钙钛矿阳离子空位态密度 | 【朱老师讲DFT计算】
VASP钙钛矿态密度计算 | 【朱老师讲DFT计算】
局域态密度作图与分析 | 态密度计算与应用-DFT计算特训营【朱老师讲VASP】
Ni表面吸附H差分电荷密度计算 | 【朱老师讲DFT计算】
Gimp和Inkscape软件的常用功能:图片裁剪与图片排版 | DFT计算特训营【朱老师讲VASP】
VASP公开课——一节课入门催化与吸附计算
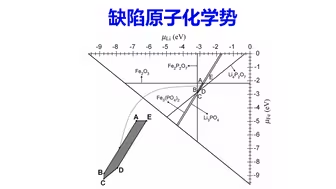
缺陷计算:半导体缺陷原子化学势基本概念 | 【华算科技朱老师讲DFT-VASP】
磁性计算:铁磁构型能量计算 | 【华算科技朱老师讲DFT-VASP】
Ni表面吸附H2O结合能 | 【朱老师讲DFT计算】
结构优化:INCAR、KPOINTS、POTCAR文件简介 | DFT计算特训营【朱老师讲VASP】
N掺杂C负载Fe单原子吸附NH3吸附能 | 【朱老师讲DFT计算】
VESTA软件功能:模型构建、理论XRD、电荷密度绘图 | DFT计算特训营【朱老师讲VASP】
缺陷形成能 |【朱老师讲DFT计算】
钙钛矿计算模型:阳离子MA | DFT计算特训营【朱老师讲VASP】
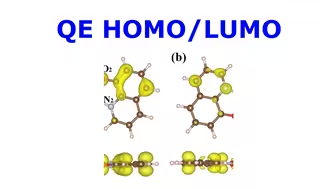
QE HOMO LUMO计算 | 【华算科技朱老师讲DFT-QE】
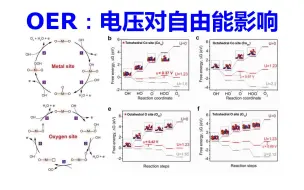
催化OER计算:电压对自由能影响 | 【华算科技朱老师讲DFT-VASP】
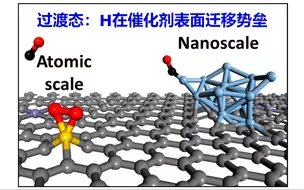
过渡态计算H在催化剂表面迁移路径与势垒 | DFT计算特训营【朱老师讲VASP】
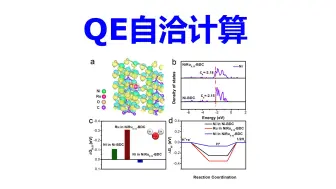
QE自洽计算 | 【华算科技朱老师讲DFT-QE】
态密度数据处理与下载 | 态密度计算与应用-DFT计算特训营【朱老师讲VASP】
VASP软件:二维材料石墨烯VASP计算N掺杂电荷密度分析 | 纯小白入门DFT计算【朱老师讲VASP】
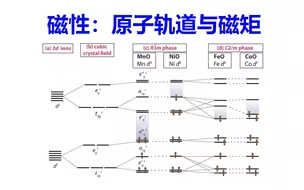
磁性计算:原子轨道与磁矩 | 【华算科技朱老师讲DFT-VASP】
金属材料结构与弹性性质计算:层错能、表面能、弹性常数计算 | DFT计算特训营【朱老师讲VASP】
磁性计算:电荷分布计算 | 【华算科技朱老师讲DFT-VASP】
VASP软件:二维材料石墨烯VASP计算石墨氮掺杂模型 | 纯小白入门DFT计算【朱老师讲VASP】